Proteogenomic-based method for identifying tumor-specific antigens
A tumor-specific, proteomic technology, applied in the field of tumor antigen identification, can solve the problems of not being able to identify TSA, not using aeTSA for high-throughput identification, and not considering two key factors.
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image


Examples
Embodiment 1
[0228] Example 1: Materials and methods
[0229] Mice. C57BL / 6 mice were obtained from Jackson Laboratories (Bar Harbor, ME). Mice were housed under specific pathogen-free conditions.
[0230] Cell lines. The EL4 T-lymphoblastic lymphoma cell line, CT26 colorectal carcinoma cell line and B-cell hybridoma HB-124 were obtained from the American Type Culture Collection (ATCC). EL4 and CT26 cells were cultured in RPMI 1640 / HEPES supplemented with 10% heat-inactivated fetal calf serum, 1% L-glutamine and 1% penicillin-streptomycin. Cell culture medium was further supplemented with 1% non-essential amino acids and 1% sodium pyruvate or only 1% sodium pyruvate for EL4 and CT26 cells, respectively. For the production of anti-CDR2 antibodies, HB-124 cells were cultured in IMDM supplemented with 10% heat-inactivated fetal bovine serum. Unless otherwise stated, all reagents were purchased from
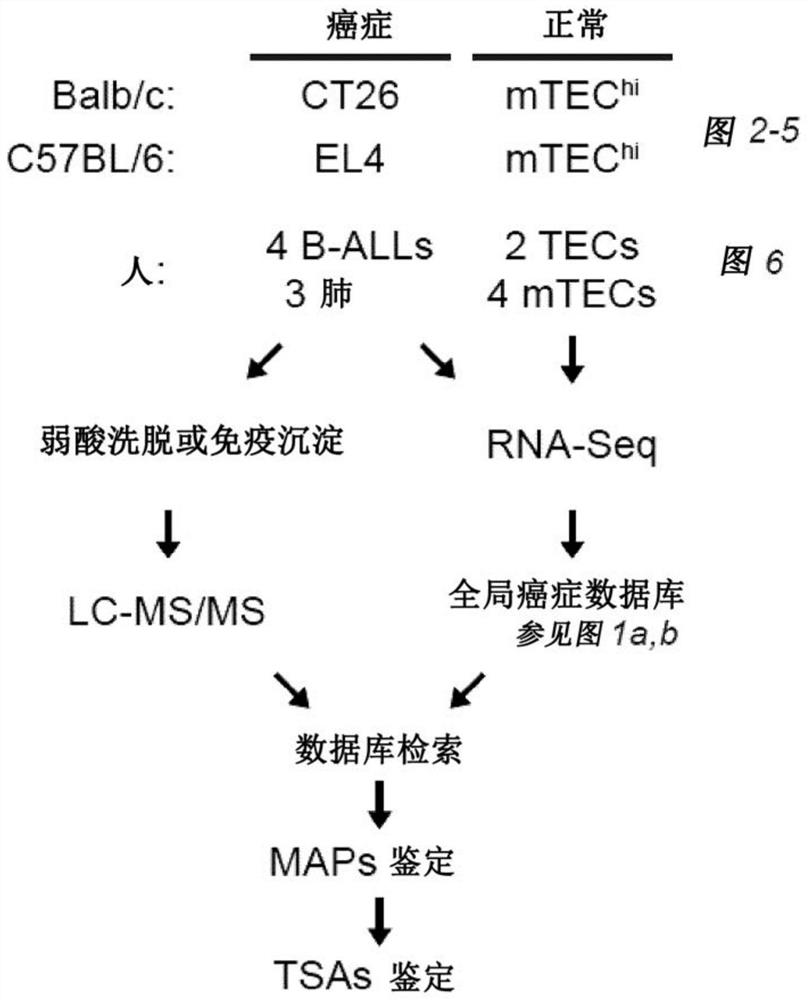
[0231]Human primary samples. The primary leukemia samples used in this study (four B-AL...
Embodiment 2
[0250] Example 2: Rationale and design of a proteomics approach for TSA discovery.
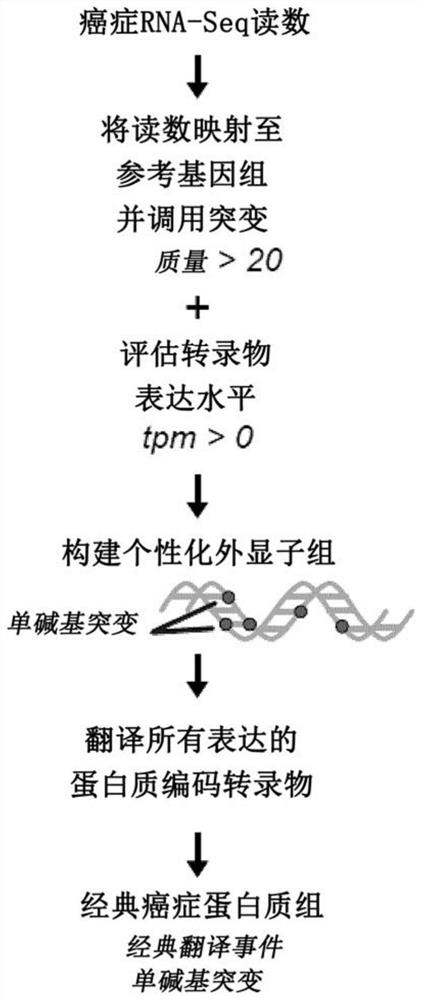
[0251] Attempts to computationally predict TSA using various algorithms are fraught with extremely high false discovery rates 27 . Thus, system-level molecular definition of MAP libraries is only possible through high-throughput MS studies 3 . Current methods use MS / MS software tools such as Peaks28 , which relies on a user-defined protein database to match each acquired MS / MS spectrum and peptide sequence. Since the reference proteome does not contain TSAs, MS-based TSA discovery workflows must use proteomics strategies to build customized databases derived from tumor RNA-sequencing (RNA-Seq) data 29 , the database should ideally contain all proteins expressed in the tumor sample under consideration, even unannotated proteins. Due to the inability of current MS / MS software tools to process full-frame translation of all RNA-Seq reads 30,31 Given the huge search space created, proteomic st...
Embodiment 3
[0252] Example 3: Noncoding regions are the main source of TSA.
[0253] At a false discovery rate of 5%, 1,875 MAPs on CT26 cells and 783 MAPs on EL4 cells were identified. Among those, if (i) their 33 nucleotide long MAP coding sequence (MCS) derived from the pan-cancer-restricted 33 nucleotide long k-mer is absent in the mTEChi transcriptome or if ( ii) Their 24-to-30 nt long MCS derived from a truncated version of the cancer-restricted 33 nt long k-mer in the transcriptome of cancer compared to mTEC hi Cells overexpressed at least 10-fold ( Figure 8A ), then mTEC hi MAPs absent in the proteome were considered TSA candidates. During the MS-related validation step and assignment of genomic localization ( Figure 8B -C) Afterwards, a total of 6 mTSA candidates and 15 aeTSA candidates were obtained: 14 presented by CT26 cells and 7 presented by EL4 cells ( Figure 2A -B). MAPs that are both mutated and aberrantly expressed are included in the mTSA category. All of thes...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More - R&D
- Intellectual Property
- Life Sciences
- Materials
- Tech Scout
- Unparalleled Data Quality
- Higher Quality Content
- 60% Fewer Hallucinations
Browse by: Latest US Patents, China's latest patents, Technical Efficacy Thesaurus, Application Domain, Technology Topic, Popular Technical Reports.
© 2025 PatSnap. All rights reserved.Legal|Privacy policy|Modern Slavery Act Transparency Statement|Sitemap|About US| Contact US: help@patsnap.com